Registrazione dei dispositivi Medici
REGISTRAZIONE DEI DISPOSITIVI MEDICI
Tutti i dispositivi medici per poter accedere al mercato cinese sono sottoposti all’obbligo di approvazione “pre-market”, da parte della NMPA (National Medical Products Administration), attraverso la pratica di Registrazione dei Dispositivi Medici.
La Repubblica popolare cinese è attualmente il mercato con il tasso di crescita più rapido al mondo per i prodotti sanitari con un potenziale enorme per i produttori esteri di dispositivi medici, prodotti farmaceutici e cosmetici. Tuttavia, l’ingresso in questo mercato è complesso a causa delle stringenti normative, dei problemi di comunicazione e di un’alta concorrenza.
Il Consorzio Italasia gestisce da anni le pratiche di registrazione per conto di grandi gruppi italiani e offre la propria esperienza per aiutare la Vostra Azienda a soddisfare tutti i requisiti imposti dalle autorità cinesi per portare a termine con successo la registrazione dei Vostri prodotti.
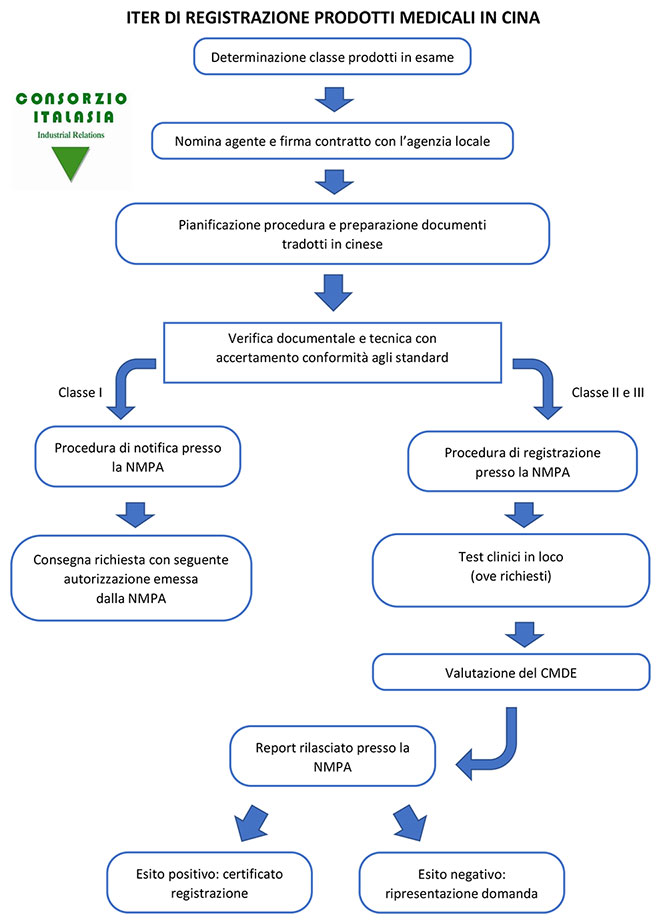
DETERMINAZIONE DELLA CLASSE DI PRODOTTI PRESI IN ESAME: I dispositivi medici vengono classificati in base a tre tipologie:
- Classe I: apparecchiature generiche non alimentate elettricamente (non attive) che non penetrano nel corpo o apparecchiature chirurgicamente non invasive per uso transitorio (meno di 60 minuti); apparecchiature alimentate elettricamente (attive) a basso rischio per uso diagnostico o di supporto del paziente. Es. strumenti operatori generici, strumenti odontoiatrici generici (pinze, forbici, ecc..), strumenti ortopedici.
- Classe II: apparecchiature attive terapeutiche e diagnostiche rischiose e non; apparecchiature chirurgicamente invasive per uso transitorio, a breve termine, a lungo termine o impiantabili. Es. cannule aspira saliva, strumenti ad ultrasuoni, aghi per agopuntura, termometri elettronici.
- Classe III: apparecchiature che entrano in contatto con il sistema circolatorio centrale del cuore o il sistema nervoso centrale; apparecchiature invasive a lungo termine o impiantabili che abbiano effetto biologico sul corpo o che siano assorbite in esso. I dispositivi di questa classe sono considerati ad alto rischio per cui la sicurezza è da testare attraverso trial clinici e di laboratorio. Es. strumenti a raggi X, aghi delle siringhe.
NOMINA DI UN’AGENZIA/AGENTE: la richiesta per la registrazione di prodotti medicali può essere condotta esclusivamente attraverso un ente legale cinese /agenzia /agente autorizzato che assisterà il fabbricante durante tutte le fasi del processo e il cui rapporto viene definito attraverso una lettera di autorizzazione. L’azienda produttrice dovrà tuttavia verificare che i prodotti vengano registrati a suo nome e non a nome dell’agente cinese, cosa che purtroppo in molto casi si verifica creando problemi di carattere burocratico/legale nel caso in cui l’azienda decida in seguito di cambiare strategia commerciale e di nominare un nuovo importatore/agente per la distribuzione dei propri prodotti nel mercato cinese.
VERIFICA DOCUMENTALE E TECNICA, CON ACCERTAMENTO DI CONFORMITA’ AGLI STANDARD: i dispositivi medici devono osservare standard nazionali e standard professionali/settoriali; se un prodotto nuovo o innovativo non appartiene ancora ad alcuno degli standard esistenti devono essere presentate le norme di produzione per la registrazione, che come minimo devono comprendere i requisiti di qualità e sicurezza degli standard nazionali e professionali. I test per comprovare la conformità del prodotto vengono effettuati da un laboratorio cinese, che viene scelto dal produttore in base ad una lista fornitagli dalla NMPA. Il laboratorio che effettua i test entra in contatto diretto col produttore/agente per richiedere le documentazioni, i campioni e i processi effettuati, dopodiché a test completati e tasse pagate, sarà cura del laboratorio emettere un rapporto provvisorio utile a verificare lo status della certificazione.
TEST CLINICI: in base alla tipologia di prodotto il laboratorio convenzionato con la NMPA chiederà una certa quantità di campioni al fine di essere testati; questi test sono obbligatori per le classi di prodotto II e III (con eventuali esenzioni).
CONSEGNA DELLA DOMANDA DI REGISTRAZIONE: una volta che il produttore avrà ottenuto una relazione positiva su test e analisi cliniche si potrà inviare la domanda alla NMPA compilando l’Application Form in cinese.
VALUTAZIONE DEL CMDE: Il Centro Cinese per la Valutazione dei Dispositivi Medici (CMDE) che fa capo alla NMPA, esaminerà i documenti e deciderà se approvare o no la domanda. In caso positivo procederà ad una serie di ulteriori accertamenti tecnici al fine di elaborare una relazione che motiva il giudizio positivo sul prodotto/apparecchiatura.
OTTENIMENTO DEL CERTIFICATO: l’approvazione finale della registrazione viene rilasciata dalla NMPA. Per i prodotti di classe I la validità della notifica è illimitata. Per i prodotti di classe II e III il certificato è valido 5 anni ed è consigliato il rinnovo 6 mesi prima della sua scadenza.
AGGIORNAMENTI GOVERNATIVI PER LA DOMANDA DI REGISTRAZIONE: In caso di esito negativo della registrazione, il governo cinese ha introdotto recentemente una serie di semplificazioni. L’Announcement n.16 del 2020 informa che il CMDE, durante l’esame e la revisione della documentazione, possa presentare suggerimenti e correzioni, in modo da permettere al richiedente di ripresentare la domanda rispettando le indicazioni fornite, così che gli venga accettata la volta seguente.
L’Announcement n.17 invece semplifica le procedure di ritiro e ripresentazione delle domande di registrazione per i prodotti presentati di classe II, giudicati poi di classe III post revisione tecnica. Il CMDE deve fornire dei documenti che spieghino i motivi di tale esclusione. Tali documenti dovranno poi essere inclusi nella successiva application per la classe corretta.
DURATA COMPLESSIVA DEL PROCESSO DI REGISTRAZIONE DEI DISPOSITIVI MEDICI: In caso di esito negativo della registrazione, il governo cinese ha introdotto recentemente una serie di semplificazioni. L’Announcement n.16 del 2020 informa che il CMDE, durante l’esame e la revisione della documentazione, possa presentare suggerimenti e correzioni, in modo da permettere al richiedente di ripresentare la domanda rispettando le indicazioni fornite, così che gli venga accettata la volta seguente.
L’Announcement n.17 invece semplifica le procedure di ritiro e ripresentazione delle domande di registrazione per i prodotti presentati di classe II, giudicati poi di classe III post revisione tecnica. Il CMDE deve fornire dei documenti che spieghino i motivi di tale esclusione. Tali documenti dovranno poi essere inclusi nella successiva application per la classe corretta.
- DISPOSITIVI CLASSE I : le tempistiche variano da 1 a 4 SETTIMANE
- DISPOSITIVI CLASSE II e III : le tempistiche variano dai 12 ai 22 MESI
Per informazioni e preventivi contattare il Consorzio Italasia di Torino:
Dott.ssa Anna Bassano
Tel. 0115627180
E-mail : bassano@italasia.it